Rows: 1141 Columns: 8
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (5): sample_type, timing, plot, location, treatment
dbl (3): site, ak_content, year
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 576 Columns: 8
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (5): sample_type, timing, plot, location, treatment
dbl (3): site, dryweight, year
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
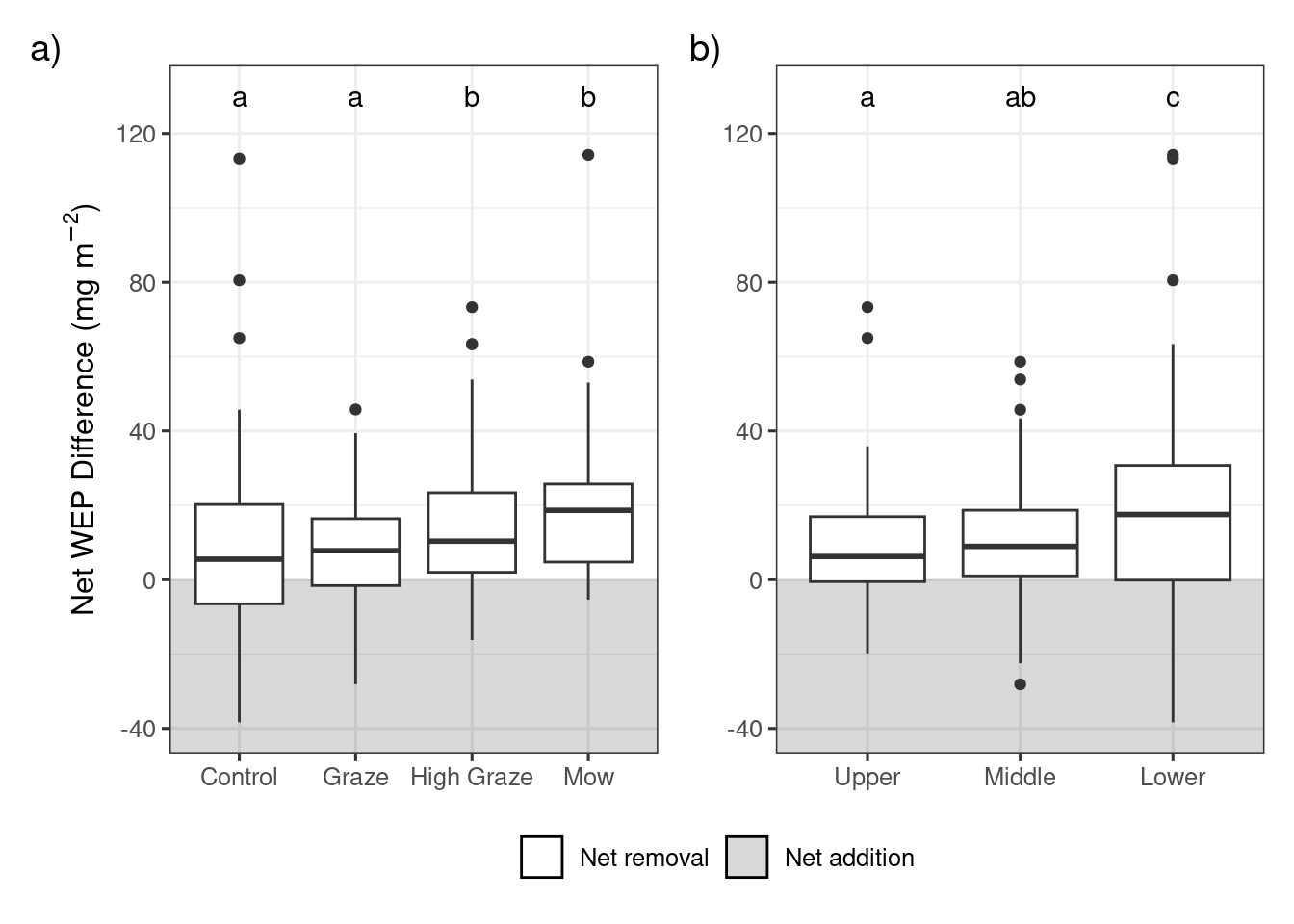
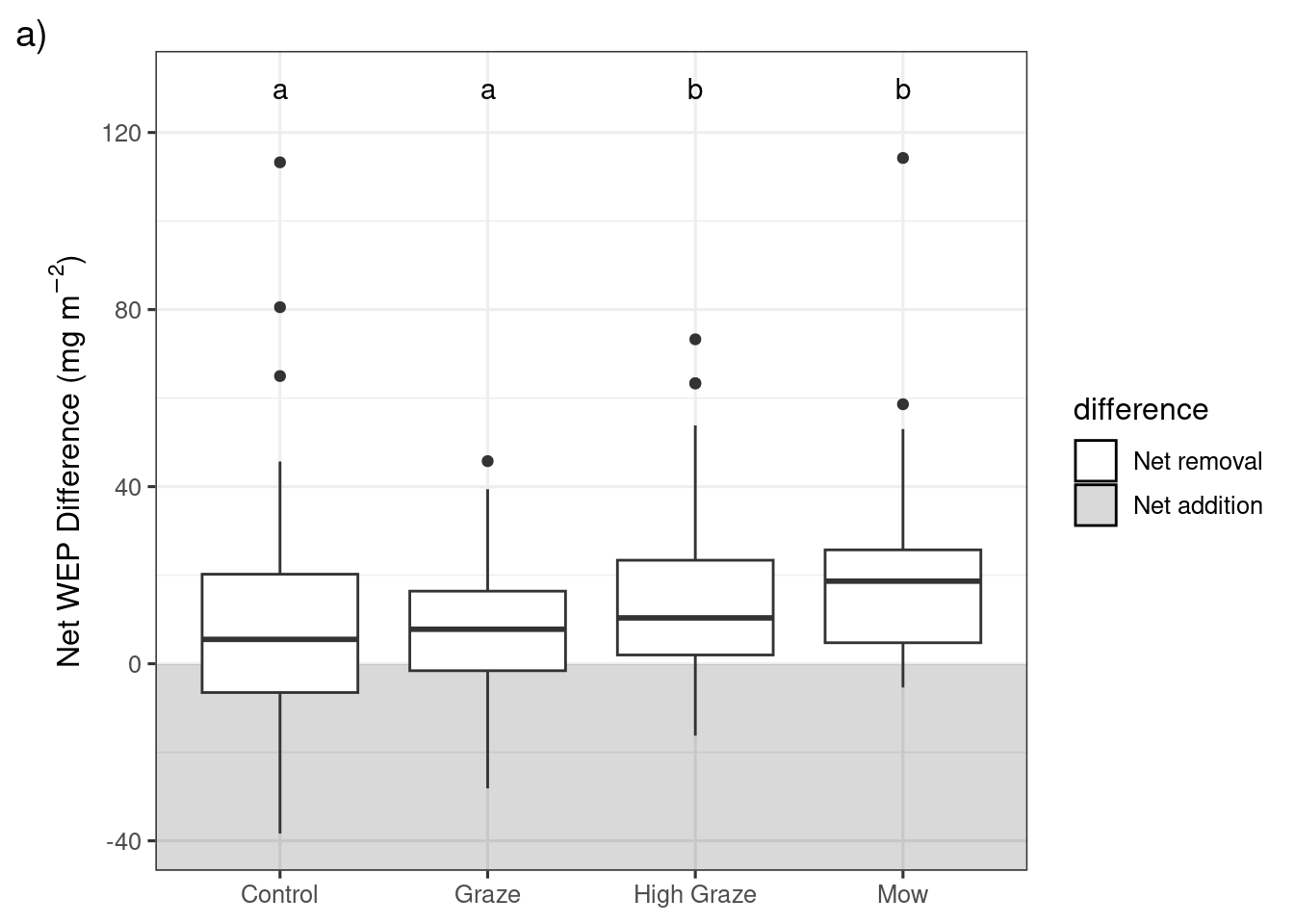
Results of the post-hoc pairwise comparisons with a Benjamini-Hochberg p value adjustment for differences in the net biomass WEP (\(mg~m^{-2}\)) between the four treatments and three riparian sampling locations.
Change in riparian biomass WEP following grazing or mowing in each riparian location. Within each plot significant differences (p<0.05) between treatments or riparian locations are denoted with different letters. Lower sampling locations are adjacent to the edge of the waterbody and Upper locations are adjacent to the field.
Dry Weight
In [15]:
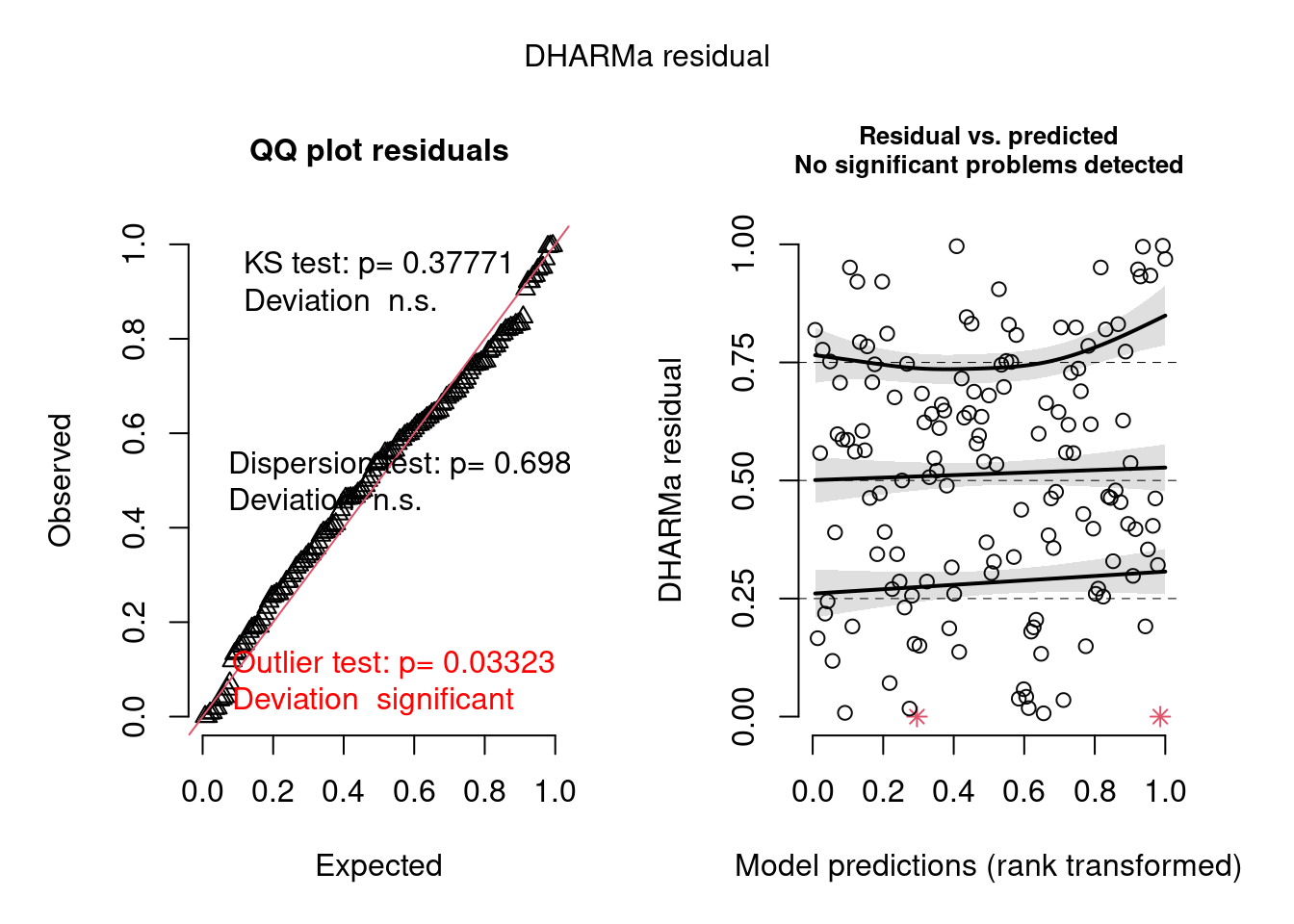
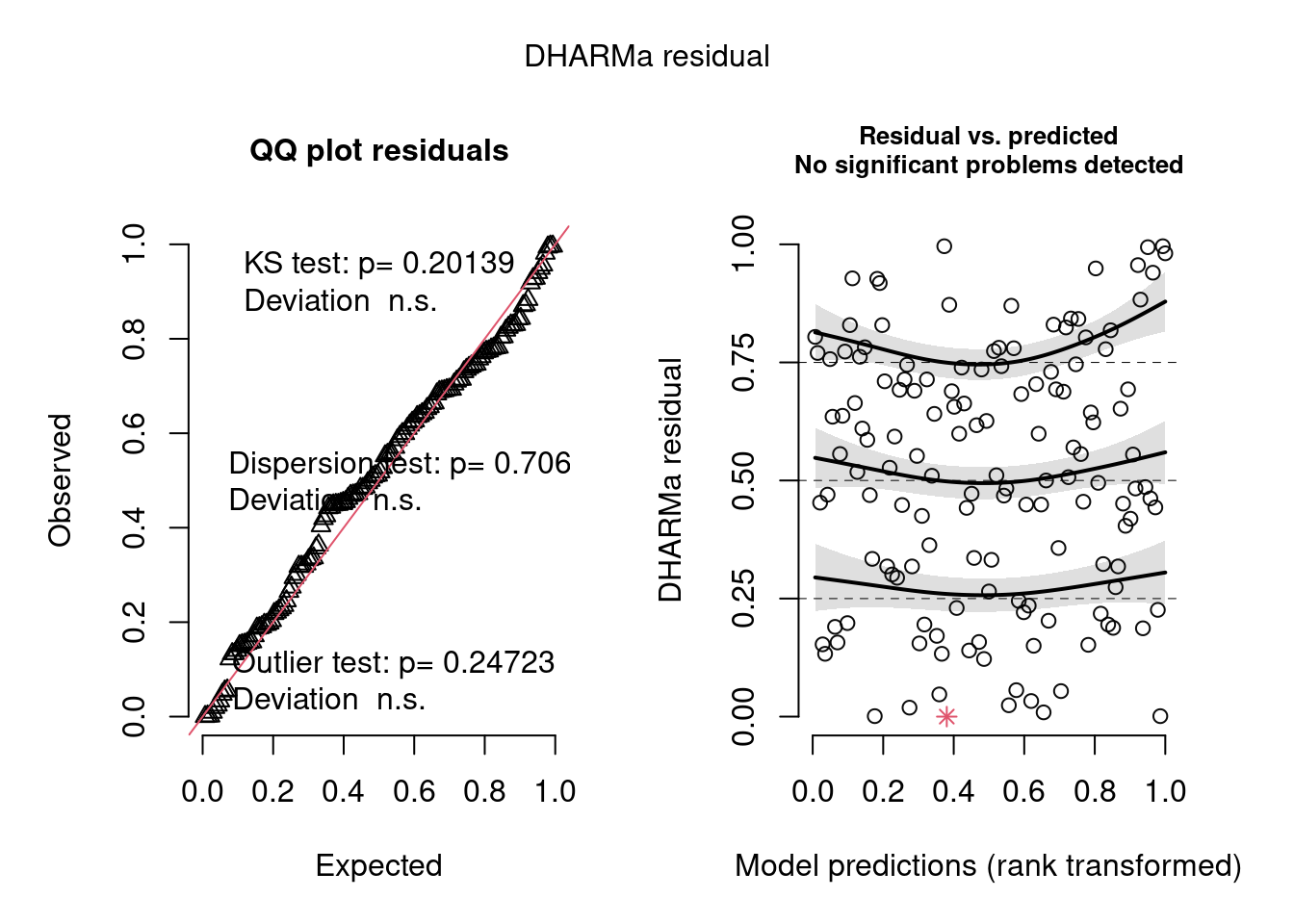
m2 <-glmmTMB((diff) ~ treatment * location + Before + (1|site) + (1|year),data =filter(biomass_diff, measure =="dryweight"))simulateResiduals(m2, n =1000, plot =TRUE)
Object of Class DHARMa with simulated residuals based on 1000 simulations with refit = FALSE . See ?DHARMa::simulateResiduals for help.
Scaled residual values: 0.854 0.648 0.326 0.646 0.318 0.725 0.668 0.466 0.81 0.586 0.427 0.882 0.777 0.897 0.805 0.368 0.704 0.594 0.586 0.522 ...